




La mise au point d’un médicament est un processus de développement long et complexe. Avant de proposer de nouveaux traitements à tous les patients concernés, il faut s’assurer qu’ils sont efficaces et bien tolérés. Le nouveau médicament est d’abord développé dans un laboratoire, puis testé sur les animaux – c’est la recherche préclinique. Si les résultats de ces tests sont favorables, il est alors possible de proposer à des personnes atteintes du cancer (pour les essais promus par Unicancer) de participer, avec leur accord, à son évaluation. Cette évaluation s’appelle un essai clinique.
L’objectif des essais cliniques
Les essais cliniques peuvent évaluer :
- de nouveaux médicaments ou associations de médicaments (pour soigner une maladie, atténuer les effets secondaires, améliorer la qualité de vie, etc.)
- de nouvelles façons d’administrer des traitements (par comprimés plutôt que par injection, etc.)
- de nouvelles techniques de traitement (nouveau type d’opération chirurgicale ou de radiothérapie, etc.)
- de nouvelles techniques de diagnostic (nouveau test biologique, etc.)
Le déroulement d’un essai clinique
Pour garantir la sécurité des patients et la rigueur scientifique, les essais cliniques comprennent 4 phases qui sont destinées à recueillir des informations spécifiques sur le nouveau traitement.
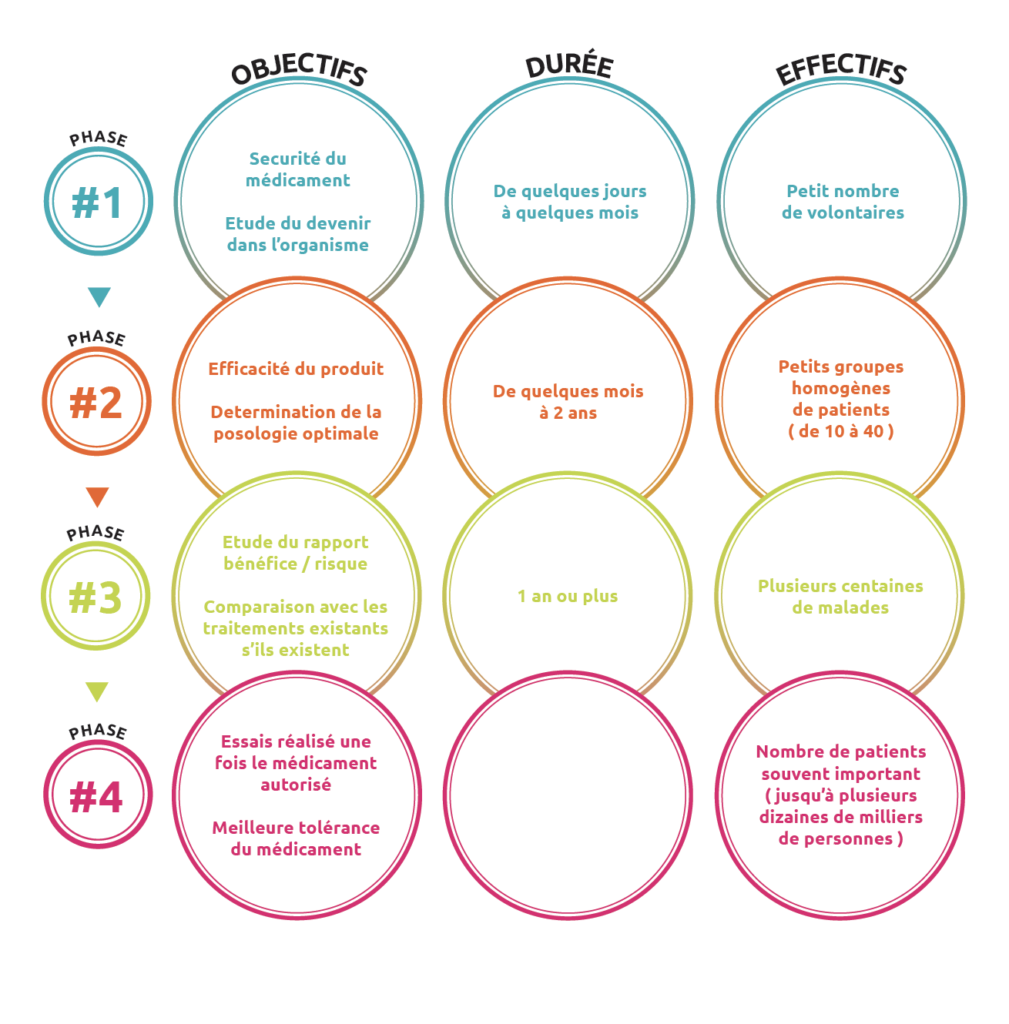
Description de l’infographie
Les essais de Phase I ont pour objectif d’évaluer la tolérance d’un nouveau médicament dans le but de déterminer la dose recommandée en terme de sécurité, et dans certains cas de donner les premières indications sur l’efficacité. Le traitement évalué est administré à un petit nombre de patients (10 à 40).
Les essais de Phase II évaluent l’efficacité d’un traitement. Ils nécessitent en général l’inclusion d’un plus grand nombre de patients (40 à 80) pour exclure les risques de conclusions erronées sur l’efficacité du traitement. Cette phase permet également de confirmer et consolider les connaissances sur la sécurité du traitement.
Les essais de Phase III sont des essais comparatifs. Ils permettent de comparer le nouveau traitement avec le traitement utilisé habituellement, dit « traitement standard ». Deux groupes de patients, homogènes et comparables (âge, sexe, caractéristiques de la maladie, etc…) sont constitués par tirage au sort (randomisation) : un groupe recevra le traitement de référence, l’autre le nouveau traitement. Ce n’est donc pas le médecin qui décide de l’attribution de l’un ou l’autre des traitements à son patient. Ces essais nécessitent l’inclusion d’un grand nombre de malades (plusieurs centaines) pour établir une réelle différence entre les traitements. Si les données et résultats de ces essais sont en faveur du nouveau traitement, cela permet de constituer un dossier d’enregistrement qui sera soumis aux autorités de santé afin qu’elles délivrent l’autorisation de mise sur le marché (AMM), ce qui autorise la commercialisation du nouveau traitement.
Quand le médicament est commercialisé, il fait encore l’objet d’une surveillance étroite appelée pharmacovigilance (Phase IV). Ainsi, tout signe anormal inattendu dû à son administration fait l’objet d’une déclaration à l’Agence Nationale de Sécurité Sanitaire des Produits de Santé (ANSM).
Le patient ne participe pas à toutes les phases de manière successive mais se voit proposer de participer à un essai dans l’une des quatre phases, en fonction de critères très précis.
Un suivi intensifié des patients durant les essais cliniques
Les patients qui entrent dans un essai clinique bénéficient d’un suivi particulier prédéfini par les modalités de l’essai, afin de vérifier l’efficacité et la tolérance du traitement. Le patient reçoit une prise en charge spécialisée avec des visites de suivi plus rapprochées et des analyses supplémentaires comparée à la prise en charge clinique habituellement réalisée pour le suivi des patients hors essai clinique.
Un cadre réglementaire bien établi, garant de la sécurité des patients
Les essais sont soumis à deux organismes de contrôle :
- Le Comité de Protection des Personnes (CPP) est un comité indépendant composé de personnes qualifiées en matière de recherche biomédicale, en éthique : professionnels de la santé (médecins, pharmaciens, infirmiers), épidémiologistes, psychologues, juristes, représentants des associations de malades et d’usagers du système de santé.
Le CPP évalue si l’essai est acceptable sur le plan scientifique et éthique, notamment la protection des patients impliqués dans l’essai. Le CPP se penche en particulier sur la sécurité physique et psychologique des patients en évaluant le rapport bénéfice/risque, mais aussi sur l’information reçue par les patients avant d’accepter de participer à l’essai ainsi que sur les mesures mises en place pour protéger les données personnelles de ces patients. - L’Agence Nationale de Sécurité du Médicament (ANSM) évalue la sécurité, la qualité et le bon usage des produits de santé utilisés pendant l’essai, garantissant ainsi la sécurité physique des patients impliqué dans l’essai.
Les deux comités doivent donner leur accord pour la conduite d’un essai clinique. Si l’un d’eux considère que les conditions évaluées ne sont pas acceptables, ils sont en mesure de s’opposer à la tenue de l’essai.